
{kind=link}

BY COMPANIES
Article 117 of the MDR introduced significant new requirements for IDDC products and the Companies making Marketing Application for these types of Medicinal Product. A117 requires a demonstration of conformity of the device component to the MDR Annex I, General Safety and Performance Requirements, either through the use of a CE certified device, self-declaration or the provision of a Notified Body Opinion (NBOp) Report.
The requirement that the device part of these IDDCs meet the relevant requirements of Annex I of the Medical Device legisation however is not a new requirement. Pre-MDR, the Medical Device Directive 93/42/EEC explicitly required for IDDCs compliance of the device part to the Annex I Essential Requirements of 93/42/EEC, however the level of information required and assessment by the Competent Authorities was not explicity stated with divergences in approach seen.
As the technology for the device part of these IDDCs becomes more complex, with on-body dosing systems and use of software to time the dosing regimens, it did become apparent that the skill set required to evaluate the safety of performance has increased, hence under MDR the introduction of the Notified Body to the process.
Data from NBCG-Med from April 2024, shown at NBCG-Med meetings and EMA meetings but unpublished, shows that 17 out of 39 notified bodies that responded have issued an Article 117 opinion (NBOp) to date (see Figure 1). As a full scope NB (1), BSI has issued over 150 NBOps since their first opinion issued in January 2020.
While the A117 Assessment process within NBs has become fully implemented and embedded within internal systems and procedures, for many pharmaceutical companies it may still be seen as a new and daunting process as the requirement to obtain a NBOp for affected medicinal products has only applied to New Marketing Authorisation Applications since May 2021.
A117 requires that Marketing Authorisation Applicants provide evidence of the conformity of the device part to MDR Annex I GSPRs. While the concept of GSPRs may be new to such applicants the following guiding principles should be considered:
- Data should be provided in Technical Documentation format, the contents of which are provided in Annex II of the MDR.
- Not all GSPRs may be applicable to the device parts for which application is made but a statement of applicability or not with a justification for any deemed not applicable is required.
- There are some GSPRs which shall have some overlap with the Competent Authority assessment, such as Stability, however differing perspectives are taken in the assessments, the NB shall want to ensure the device part can perform over the shelf-life of the product while the Competent Authority assessment is concerned with the chemical stability of the medicinal product over the proposed shelf-life.
- As the final integral drug device combination is regulated as a medicinal product, the labelling needs to conform to the requirements of the Medicinal Product Directive, however where labelling solutions have been implemented as part of risk mitigation, the NB review shall include an assessment of these aspects.
From experience of conducting many NBOp assessments, the evidence used to demonstrate compliance to GSPRs may be sourced from many sources such as literature, suppliers and sub-contractors, in addition to in-house data.
Information from unpublished EMA data suggest 25% of Market Authorisation Applications include an IDDC. Over the period reviewed, 68 IDDC applications were made. Out of these 68 procedures, 75% had one or more NBOps currently available. The others are still to be provided prior to Committee for Medicinal Products for Human Use (CHMP) opinion. Only two Market Authorisation Applications provided a Declaration of Conformity (DoC) in place of a NBOp.
One of the issues raised by EMA when reviewing applications echoes feedback from the pharmaceutical industry and relates to the issue of Classification of IDDCs. Article 117 allows manufacturers of Class I devices (or to be precise, device parts of IDDCs) to present a DoC as part of their market authorisation application. For other classes, a CE certificate (see below) or a NBOp is required. There are no additional assessment requirements stated for higher risk classified device parts as part of Article 117 and in addition, the Classification Rules set out in Annex VIII of the Medical Device Regulation (EU) 2017/745 are designed to classify medical devices, not integral device parts of medicinal products. The classification rules are useful in understanding the risks associated with IDDCs, for example, the degree of invasiveness, inclusion of software, the utilisation of animal tissue etc; however, beyond that accurate categorisation is not required. With respect to qualification and classification the questions to be asked are: Does the product meet the definition of an IDDC (Article 1, Section 8 second paragraph or Article 1, Section 9 second paragraph)? Is a NBOp required? As mentioned for all but Class I devices a NBOp is required unless a CE certificate is included. We are not aware of the latter condition being used, most likely as it is not expected an integral device part has a standalone certificate. Even if a CE marked device were used as part of an IDDC would the scope of the certificate and the Declaration of Conformity that sits behind it cover the combined integral product? For these reasons, the NBOp is the most common way to demonstrate conformity to the Annex I requirements of the MDR.
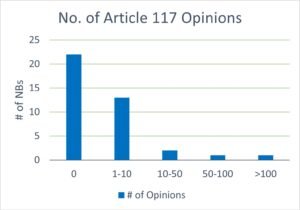
Figure 1: Number of Article 117 opinions issued by EU Notified Bodies.
Of the NBOps seen by EMA approximately 20% concerned line extensions. The majority submitted were initial NBOps, with a limited number of NBOP changes seen. The management of changes remains an area of debate and concern for Industry. There has been a lot of discussion at conferences seeking additional prescriptive guidance on the threshold for changes, or variations to an IDDC, with respect to the need for a new or updated NBOp. Theoretically, changes to the device part of a IDDC licenced prior to May 2021 would require a full new NBOp as these products would not have been previously assessed by the notified bodies. For a product authorised after this date, as an initial NBOp was provided as part of the MA Application, an updated NBOp limited to the assessment of the impact of the change to the GSPRs could be presented.
There is concern the timeline for a change to a NBOp or a new NBOp is likely to be longer than the variation review timelines for the pharmaceutical part of the device. As not many significant changes to the device part of an IDDC, that require a NBOp as part of the variation, have been seen to date, it is difficult to know the impact of this requirement. Guidance on the impact of changes on the GSPRs has been provided by Team-NB (2). In addition, EMA discuss the impact of changes to IDDC on applications in their Q&A (3). This guidance can be used to help the manufacturer assess the impact of a proposed variation on the device parts and therefore the need for an updated NBOp.
Other guidance has been produced including guidance on the documentation and timelines required by the notified bodies (4). As might be expected, the more comprehensive the documentation, the faster the review. This includes documentation from suppliers, for example where off the shelf device parts are used, the supplier’s documentation of GSPR compliance is a useful piece of the conformity puzzle. Pharmaceutical manufacturers that are proactive and prepare this information in advance through suitable relationships with these suppliers and the appropriate agreements will find faster review times.
Review times differ between NBs and are dependent on the quality of the documentation provided and number of review rounds needed to close out any open points. Assuming the manufacturer has the evidence to support compliance to the relevant GSPRs, an opinion typically takes between 2 and 6 months to be issued. Therefore, with suitable planning, including allowing time for contractual negotiations, the NBOp should be available at the start of the Marketing Authorisation Application assessment. Early dialogue and planning with the NB is a key step in ensuring CA timelines can be met.
References
- [Online]
- Team-NB. Position paper for the interpretation of device related changes in relation to a Notified Body Opinion as required under Article 117 of Medical Device Regulation (EU)2017/745. [Online] 2020.
- Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies with respect to the implementation of the Regulations on medical devices and in vitro diagnostic medical devices (Regulations (EU) 2017/745). [Online]
- MDR Article 117 Drug-device combination products application process. [Online]
Previous article:
Continue the reading:
- Device-Drug Combinations and MDR Rule 14
- Substance-Based Medical Devices – MDR Rule 21
- Borderline Cases & Classification Disputes