
{kind=link}

Nell’ambito delle varie analisi che ISPE/GAMP Italia Gruppo di lavoro «Electronic Signature» ha portato avanti per approfondire le tematiche e, anche le problematiche, annesse all’utilizzo delle firme elettroniche nel mondo farmaceutico, è stato costituito un sottogruppo che si è focalizzato sulla valutazione dei dati critici all’interno del processo GMP e sui documenti che richiedono una firma elettronica.
Infatti, nelle varie discussioni affrontate ci si è resi conto che nonostante le normative siano già in vigore da qualche decennio, e ci riferiamo a 21 CFR Part 211, EU Vol. 4, questo è ancora un argomento bisognoso di risposte concrete.
Cosa abbiamo provato a fare?
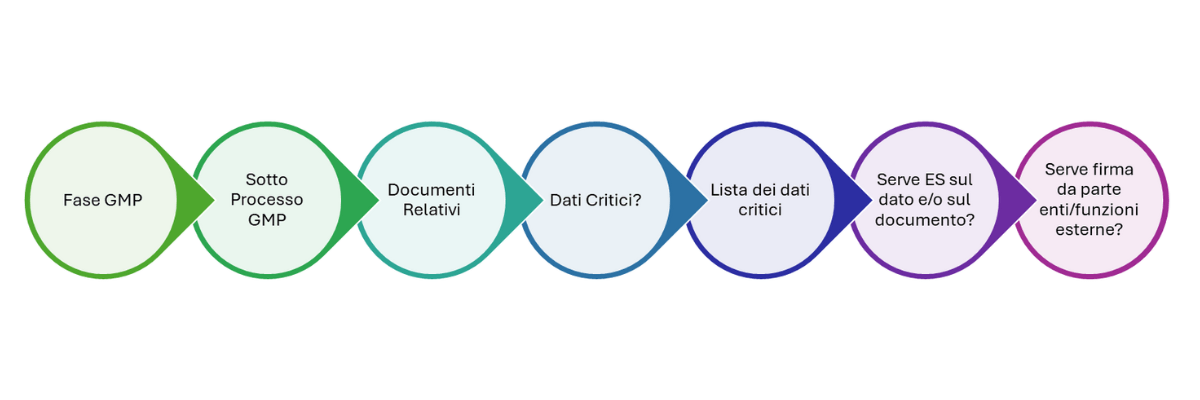
Abbiamo creato una mappatura dei processi GMP con i relativi documenti emessi, per individuare quali dati critici essi riportano e quali di questi dati sono memorizzati nei sistemi elettronici ad essi associati. La domanda che ci siamo posti è stata “cosa è necessario firmare: il dato, il documento o entrambi?”
Per farlo siamo partiti dalla fine cioè dal documento firmato all’interno del processo GMP per poi risalire ai dati critici che contiene. A questo punto il gruppo di lavoro ha cercato di darsi dei criteri per individuare quali di questi dati, che sono generati e/o processati nei sistemi, dovrebbero essere firmati elettronicamente. In primis chiedendosi dove il processo di firma ad essi applicato implica un’assunzione di responsabilità, in quanto è parte della fase decisionale che consente di andare avanti o di fermarsi, rispetto ad una semplice autenticazione dell’attività svolta. Ma andiamo con ordine.
Come anticipato, per fare la mappatura sono stati considerati i principali processi GMP ovvero: Manufacturing, Laboratorio, Manutenzione e Calibrazione, QA, Regolatorio, Qualifica e Validazione e IT/OT. Ovviamente considerata la complessità di questi macro-processi è stato necessario suddividerli nei loro sottoprocessi andando ad individuare per ciascuno le principali categorie di reportistica emessa. Quindi, per ogni tipologia di documento è stata valutata la criticità del dato gestito, ovvero dato GMP critico o non GMP critico, ad es. CPP (parametro critico di processo) CQA (attributo critico di qualità) ed è stato considerato l’impatto del dato sulla disposizione del lotto e sugli step downstream di lavorazione.
L’ultimo parametro che abbiamo analizzato è stato quello relativo al coinvolgimento di terze parti esterne nel processo di firma (es. Quality Technical Agreement stipulati con fornitori, lavorazioni per clienti, ecc..). In questo caso il coinvolgimento di terzi ha esteso l’ambito di discussione anche alle modalità della firma, alle certificazioni richieste per la sua veridicità ed alla tipologia di tool da utilizzare e ha permesso di aprire ed estendere la riflessione ad altri sottogruppi di lavoro negli ambiti GxP.
Considerazioni e alcuni esempi di processi GMP
Per quanto riguarda l’ambito GMP manufacturing, considerando il sotto-processo relativo all’esecuzione del lotto, il documento firmato è il Batch Record e, ovviamente, contiene moltissimi dati critici, fra cui ad esempio i CPP (parametri critici di processo). La prima domanda che ci si è posti è stata se questi parametri critici sono inseriti manualmente o meno nel sistema informatico. Abbiamo convenuto che se inseriti solo manualmente da parte di un operatore, la firma elettronica che ne autorizza l’utilizzo, intesa come assunzione di responsabilità per attestare che siano effettivamente quelli da utilizzare, è necessaria. Mentre se gli stessi parametri sono già parte della ricetta del sistema informatico MES o E-BR e se ci sono gli appropriati livelli di autorizzazione per modificarli (solo chi ha l’opportuno privilegio all’interno del sistema può intervenire per settarli) si è concluso che è sufficiente l’autenticazione della persona che esegue l’attività.
Nel sottoprocesso di dispensing, il documento finale è il report delle pesate. Anche in questo caso abbiamo concluso che se il sistema elettronico è interfacciato direttamente con una bilancia, non serve nessuna firma elettronica del peso della sostanza ma è necessario solo poter risalire all’autore dell’attività di pesatura. Mentre se la bilancia non è collegata al sistema elettronico allora il valore di pesatura va avallato da una firma elettronica che indica assunzione di responsabilità per il valore inserito.
Passando a un sotto-processo di monitoraggio di Temperature e Umidità di un magazzino il documento generato è un trend periodico e in questo caso i dati critici sono quelli relativi alle soglie di allarme impostate per l’analisi del trend; quindi, una firma elettronica serve quando si autorizza la modifica di tali soglie. Discorso a parte per la fase di riconoscimento di allarmi dove non sempre potrebbe essere necessaria la firma elettronica, a volte la semplice autenticazione è sufficiente a certificarne la presa visione.
Considerando il sotto-processo relativo all’esecuzione dell’analisi, in cui il certificato analitico è il documento e i risultati sono i dati critici, si deve distinguere se questi risultati vengono inseriti solo manualmente nel sistema LIMS/LES o meno. Quando inseriti solo manualmente, questi dovrebbero essere firmati dall’analista come assunzione di responsabilità dell’autenticità di quanto inserito, mentre se catturati in automatico, in quanto i sistemi LIMS/LES sono interfacciati direttamente con la strumentazione analitica, allora è sufficiente l’autenticazione della persona che ha eseguito l ‘attività. Per quanto concerne invece la parte di revisione del dato analitico, la firma è sempre necessaria perché implica un’assunzione di responsabilità a procedere con gli step a valle di rilascio e di certificazione della conformità del batch analitico.
Nel sotto-processo di campionamento di un lotto, il report generalmente contiene solo delle istruzioni operative, e non ci sono dati critici da firmare.
Anche quando si considera il sotto-processo di revisione dell’audit trail di un sistema di laboratorio, il documento generato contiene tutti dati critici ai fini della revisione, non c’è possibilità di apporre firme sul singolo metadato e, per la natura stessa del documento, non ha senso la firma elettronica del report fa parte del processo di revisione generale.
Per i processi QA, dove possono ricadere sia la preparazione di MBR che di SOP che costituiscono i documenti dei sotto-processi, tutti i dati contenuti sono critici, e non ha senso la firma del singolo dato, è il documento stesso che per sua natura ha necessità di entrare in un flusso di revisione ed approvazione omnicomprensivo.
Anche considerando il processo di incident e problem management, in ambito IT/OT network, i singoli dati che costituiscono l’analisi del problema non necessitano di apposizione di firma in quanto è l’intero framework che ha generato l’evento a costituire il report di Root Cause Analysis, che identifica i potenziali impatti sul processo di manifattura.
Nel caso di convalida e qualifica, sebbene la documentazione possa recepire raw data critici dal campo, finalizzati a certificare la corretta funzionalità del sistema rispetto all’intended use previsto (es. i risultati di una mappatura termica di una camera climatica, verifica di componenti as-built in un impianto di produzione e distribuzione di acqua purificata, ecc.) che vengono allegati, l’approvazione del report di validazione consente in maniera implicita di certificare anche la conformità dei dati in essa riportati. Nel caso si impieghino sistemi computerizzati per l’esecuzione delle attività di qualifica/convalida, la raccolta di evidenze è parte integrante del processo stesso; pertanto, l’autenticazione dell’utente che sta effettuando l’acquisizione delle evidenze può sostituire l’apposizione della firma sulle stesse.
Nel caso di processi di manutenzione, la firma del report di manutenzione, comprensivo di tutti gli interventi effettuati e del relativo esito, è sufficiente per garantire tracciabilità e conformità dell’operazione.
Ovviamente nel corso delle discussioni abbiamo anche potuto discriminare quelli che sono gli “special cases” ovvero quando, in base al tipo di sistema elettronico, a come è stato implementato e configurato, alle sue caratteristiche peculiari, al fatto che sia più o meno avanzato o nuovo e in linea con l’evolversi delle tecnologie digitali, la necessità di apporre firme elettroniche è sempre necessaria nei seguenti casi:
- quando si fanno attività critiche di data entry in manuale che il sistema elettronico non è tecnicamente il grado di gestire
- quando i sistemi che scambiano dati del processo non sono interfacciati fra loro (es. IPC)
- quando i processi non sono automatizzati.
Qualche considerazione di tipo statistico
A seguito della mappatura abbiamo provato a fare anche una valutazione statistica dei risultati ottenuti andando ad individuare per ogni sotto-processo e quindi per ogni tipologia report la % dei dati critici che un documento contiene per risalire alla % di firme elettroniche che si potrebbero apporre sul singolo record invece di generare e firmare nuovamente il documento stesso.
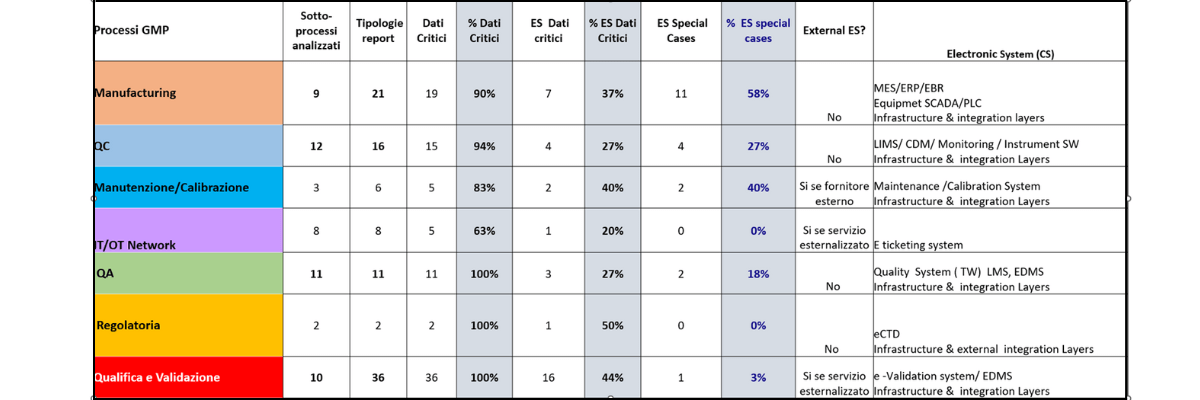
La tabella sopra dimostra che, in funzione della numerosità dei sotto-processi analizzati e della relativa reportistica, i risultati dell’analisi statistica sono da considerarsi maggiormente esaustivi per i processi di manufacturing, quality control, qualifica e validazione, per i quali si evince che:
- I sotto-processi a cui sono associati la maggior parte dei dati critici appartengono all’area qualifica/validazione, manufacturing, quality control.
- La firma elettronica su dati critici è prevista prevalentemente nei sottoprocessi afferenti all’area qualifica e validazione, manufacturing e quality control.
- Gli “special cases” dipendono dal livello di automazione del processo gestito dal sistema.
- Il coinvolgimento di terze parti esterne è generalmente previsto prevalentemente in ambito manutenzione/calibrazioni (fornitore del sistema/impianto), IT/OT network (per servizi esternalizzati o cloud) e qualifica/validazione (per attività consulenziali).
In linea generale questi risultati ottenuti hanno confermato le nostre aspettative, ovvero che i flussi di firma, che appartengono al vecchio mondo cartaceo, non devono essere applicati tal quali nei sistemi elettronici in quanto appesantiscono e rendono rigidi i flussi operativi senza dare valore aggiunto.
Inoltre, l’analisi puntuale dei processi e la valutazione della criticità dei dati si è dimostrato uno strumento essenziale per ridurre la quantità di firme apposte andando ad orientarsi verso situazioni in cui la firma ha effettivamente significato e dov’è esplicitamente richiesta da un punto di vista normativo. Tale approccio consente una standardizzazione ed un significativo efficientamento del lavoro.
Come ultimo aspetto è stata confermata la direzione di progredire verso un’analisi dei dati automatizzata, basata sulle eccezioni, in modo da concentrare lo sforzo sugli eventi significativi e in modo da prediligere un’azione proattiva e predittiva nella gestione degli eventi associati alla produzione GMP.
Riassumendo, grazie al contributo e all’esperienza dei membri di questo team, siamo riusciti a individuare le principali tipologie di dati critici che possono essere firmati elettronicamente all’interno di un sistema che gestisce i diversi processi GMP, abbiamo ovviamente constatato che esistono dei casi speciali che dipendono sia da come è stato progettato il processo sia dal livello di automazione che è stato raggiunto e convalidato.
La convalida e la gestione del dato in accordo ai principi ALCOA+ costituiscono la base per prendere le giuste decisioni.
Il lavoro svolto dal gruppo ha aperto la strada ad un’analisi approfondita anche nell’ambito dei processi GCP e GVP essendo la metodologia identificata trasversale e comune anche agli altri ambiti GxP.
Si ringraziano tutti i partecipanti del sottogruppo di lavoro per il contributo all’attività e all’elaborazione dell’articolo.